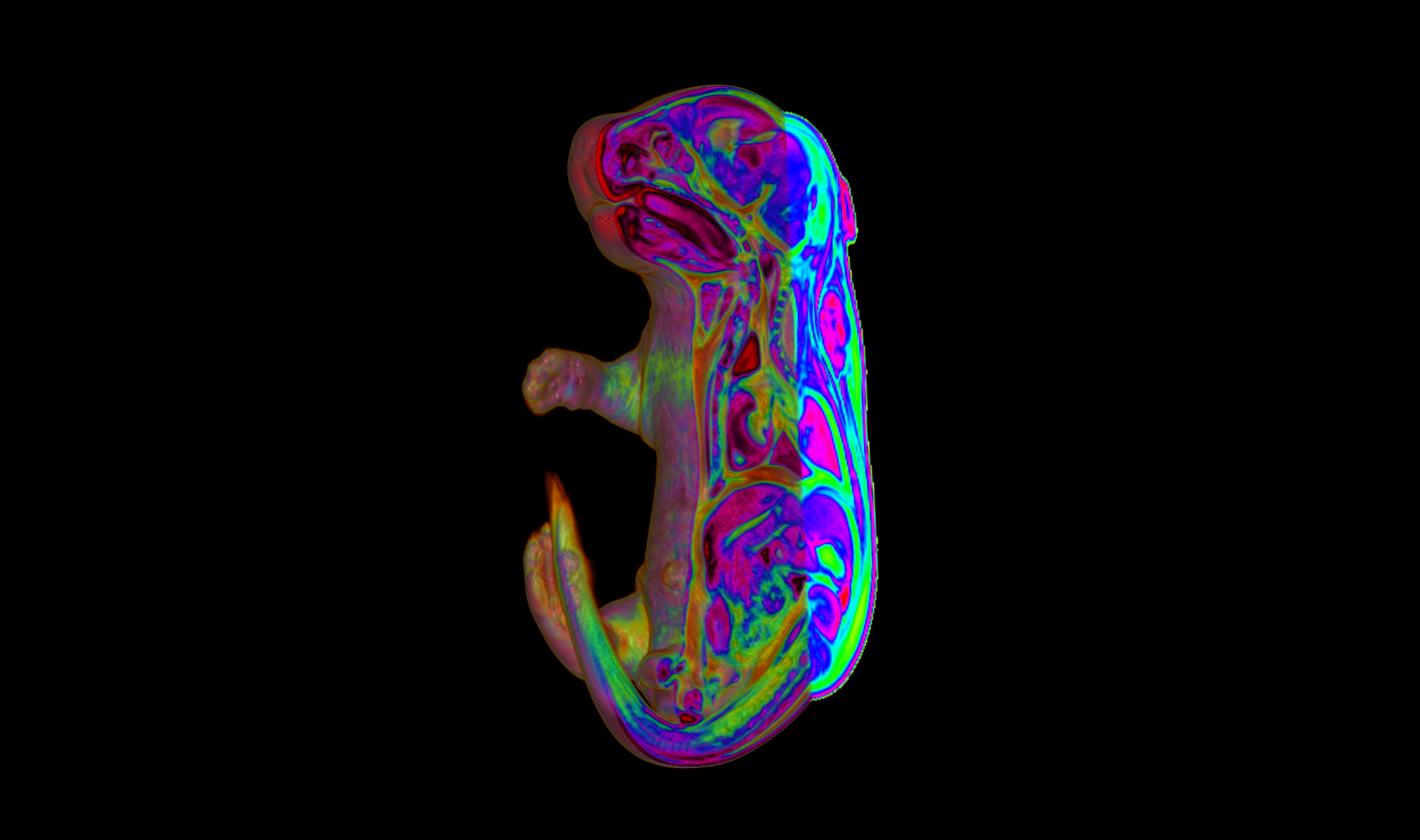
Face morphogenesis: cellular behaviors, regulatory mechanisms, epigenetic landscapes
The vertebrate head is anatomically the most sophisticated part of the body and its evolution was fundamental to the success of vertebrates on earth. The face and its features best identify us as human beings. The face conveys our thoughts and emotions, is a gateway to interpersonal communication, and a criterion for mate selection. Those who suffer from birth defects that maim facial features suffer severe hardship in society since very young age, especially when they start school.
We have acquired knowledge of the cellular behaviors, genetic pathways, regulatory and epigenetic landscapes that underpin the patterning and morphogenesis of cephalic structures, in particular the fusion of head prominences in the midface (which comprises upper lip, nose, and cheeks). Some of the most common human congenital diseases involve disfiguring abnormalities of the midface, including clefting of the lip and palate.
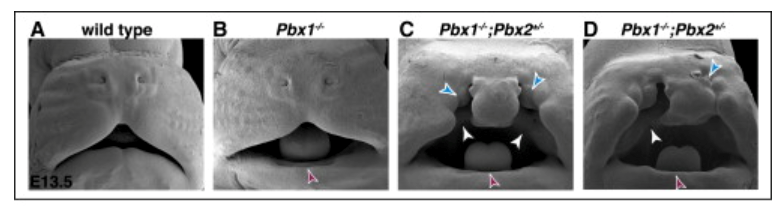
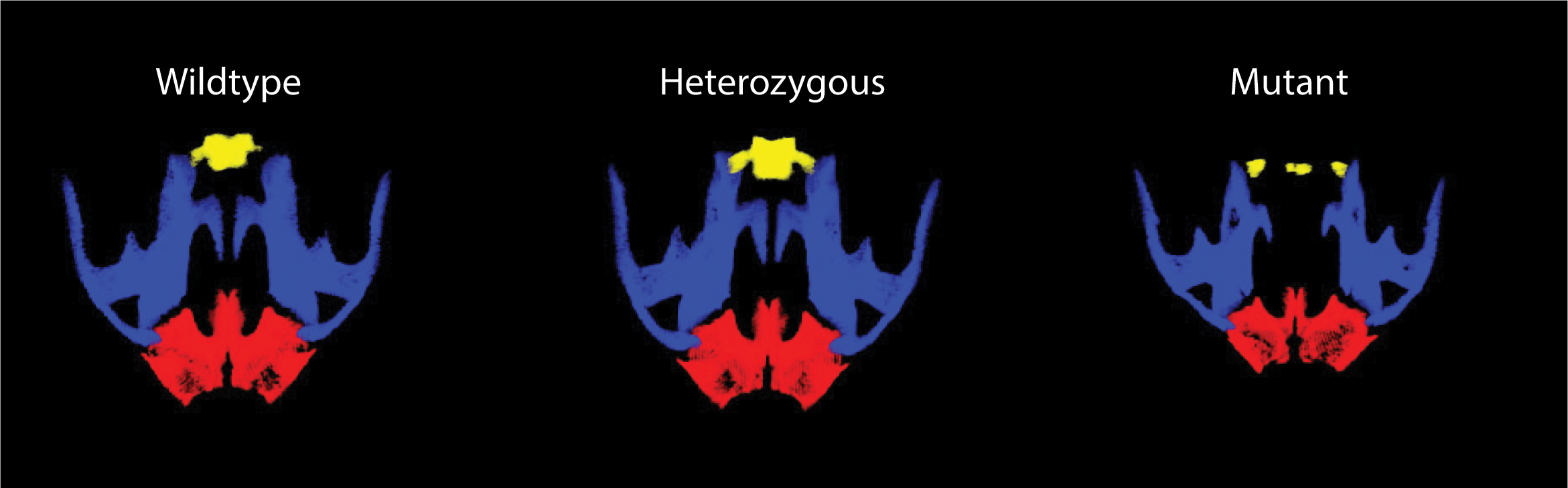
 Our studies have established the presence of novel PBX-directed, HOX-independent regulatory modules that control midfacial morphogenesis. In fact, Pbx compound mutant mice provide one of the few existing mammalian models to study the molecular basis for these defects. The perturbation of PBX-dependent regulatory modules in the midface, caused by compound loss of Pbx genes, results in localized suppression of apoptosis in the cephalic epithelium, at the seam where the frontonasal processes fuse with the maxillary process. Absence of apoptosis in Pbx mutants is accompanied by concomitant loss of EMT and tissue remodeling, all of which cause clefting of the lip and primary palate. Recently, using single-cell transcriptomics, we have identified different subpopulations within the cephalic epithelium and are uncovering for the first time how these diverse epithelial subpopulations, characterized by distinct transcript signatures, contribute to morphogenesis and fusion of the frontonasal and maxillary processes to form the beautiful face of the mouse and human embryo.
Our studies have established the presence of novel PBX-directed, HOX-independent regulatory modules that control midfacial morphogenesis. In fact, Pbx compound mutant mice provide one of the few existing mammalian models to study the molecular basis for these defects. The perturbation of PBX-dependent regulatory modules in the midface, caused by compound loss of Pbx genes, results in localized suppression of apoptosis in the cephalic epithelium, at the seam where the frontonasal processes fuse with the maxillary process. Absence of apoptosis in Pbx mutants is accompanied by concomitant loss of EMT and tissue remodeling, all of which cause clefting of the lip and primary palate. Recently, using single-cell transcriptomics, we have identified different subpopulations within the cephalic epithelium and are uncovering for the first time how these diverse epithelial subpopulations, characterized by distinct transcript signatures, contribute to morphogenesis and fusion of the frontonasal and maxillary processes to form the beautiful face of the mouse and human embryo.
Roles of the ESCRT machinery in craniofacial development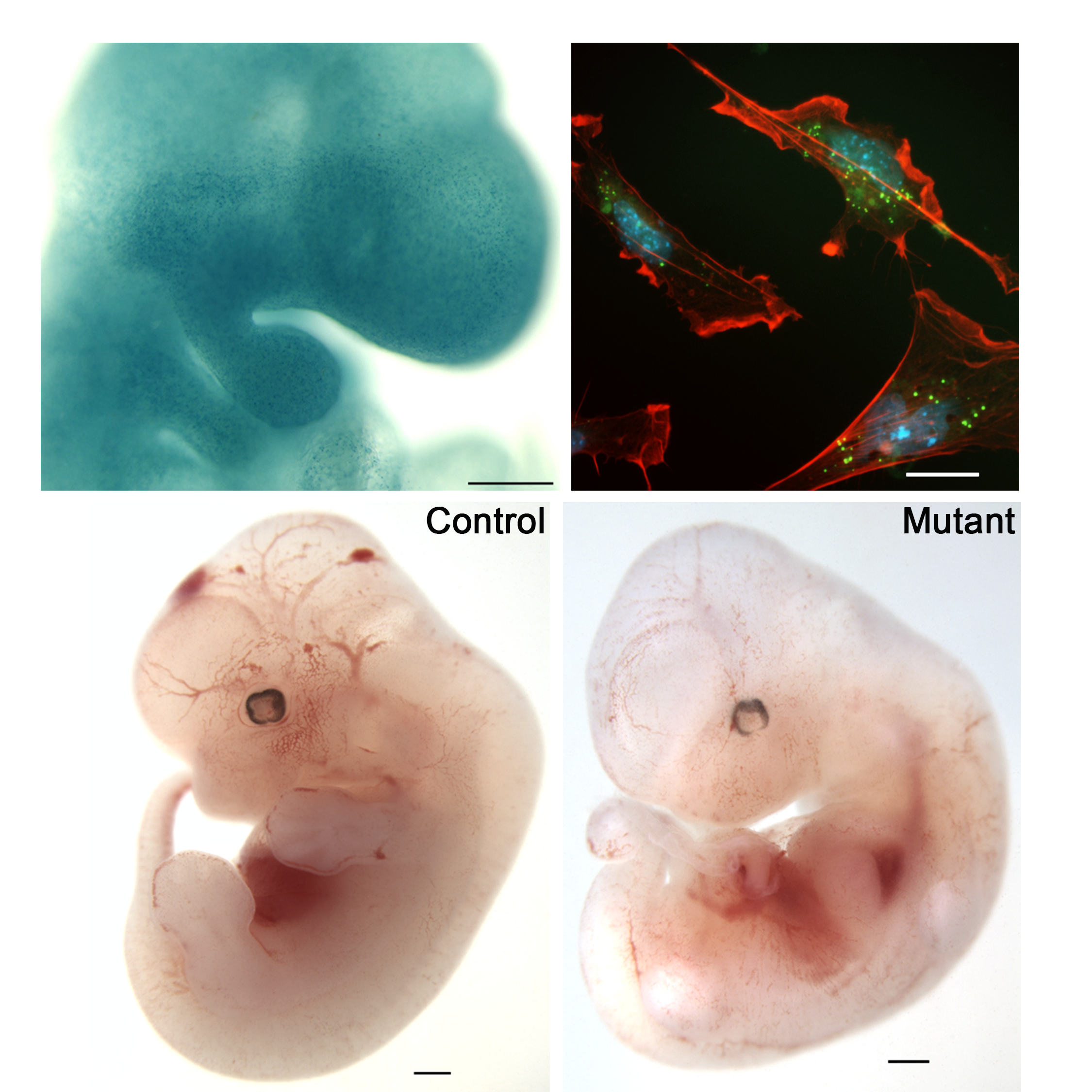
Within craniofacial development, we have identified recessive mouse mutant lines with remarkable craniofacial phenotypes, via a forward genetic screen by ethylnitrosourea (ENU) mutagenesis. We characterized one of these mouse mutant strains, exhibiting a striking hole in the temporal bone (hence we named the mutant line Hol). The defects of the cranium, coupled with the lack of pharyngeal pouch-derived structures, dysmorphic heart chambers, and limb phenotypes, led to the observation that Hol bears strong similarities to mouse models of DiGeorge and CHARGE Syndromes. We discovered that the Hol mutation affects Vps25, a gene that encodes a member of the ESCRT-II endosomal sorting machinery. Our work established that disruption of the endosomal sorting machinery is a yet unknown mechanism underlying abnormal morphogenesis of craniofacial and limb structures. We are currently characterizing additional conditional loss-of-function alleles of Vps25 and other ESCRT-encoding genes to dissect their tissue-specific roles despite their ubiquitous expression. By mass spec analysis we are also identifying new interactors of the ESCRT complex in the mammalian embryo.
Evo-devo approaches to uncover mechanisms underlying the diverse shapes of the face across species
We are fascinated by the different shapes, features, and outgrowth of the face across different species and aim to identify the genes, regulatory networks, epigenetic lanscapes, and signaling pathways that are directly involved in “making faces” in the animal kingdom. Within the Mouse ENCODE Consortium, we have gained insight into shared and species-specific transcriptional programs at the whole-genome level to identify the forces that shape mammalian regulatory DNA landscapes. Within the FaceBase Consortium, in collaboration with Joanna Wysocka and Tomek Swigut, Stanford University & HHMI, we have also generated datasets of chromatin landscapes, cis-regulatory repertoires, and transcriptomes of the human and chimpanzee cranial neural crest. This research has uncovered functionally significant cis-regulatory divergence underlying human-specific craniofacial traits .
.
By conducting comparative transcriptomics of midfacial anlagen in different species, including chick, mouse, and pig, we have recently identified a gene encoding a Zn finger- and homeobox-containing transcription factor that is expressed in embryonic midfaces of mammals, like mouse and pig, but is not expressed in the avian midface. This transcription factor is critical for midface morphogenesis and palate closure in mammals. Strikingly, when mutated, it causes orofacial clefting in both mice and humans, reminiscent of the avian palate, which is physiologically cleft.
Among the critical signaling pathways that mediate cellular and molecular programs involved in craniofacial development are those directed by calcium. Conserved involvement of calcium signaling has been implicated in craniofacial morphogenesis across species, from avian to mammal, but the mechanisms by which calcium directs the outgrowth of individual craniofacial structures are unknown. Differential expression of calmodulin (CaM), a calcium sensor, has been shown to direct midface outgrowth, specifically beak lengths, in Darwin’s finches. In humans, changes in intracellular calcium caused by a gain of function mutation in CACNA1C, which encodes the calcium channel Cav1.2, lead to Timothy Syndrome, characterized by autism, cardiac dysfunction, and craniofacial defects. Intracellular calcium elevations activate downstream signaling, such as the calcineurin (CaN)/NFAT cascade. An ongoing project in close collaboration with Georgia Panagiotakos at UCSF aims to understand the cellular and molecular mechanisms by which CaN/NFAT signaling directs midface outgrowth in species with different midfacial lengths. The small zoo we host for this project includes chicken, lizards, and mice.
Spleen morphogenesis and growth: models of human congenital asplenia
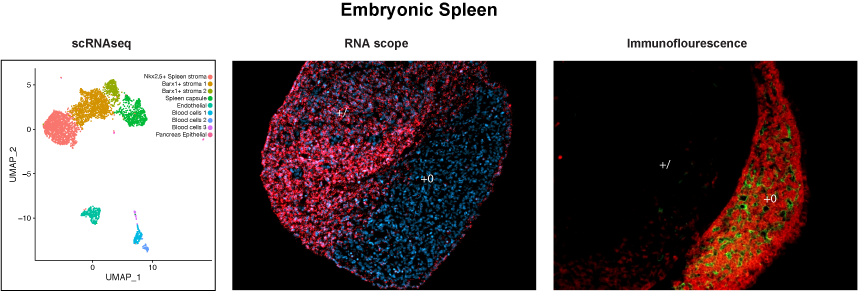 We have devoted substantial efforts to understand the genetic and transcriptional networks that control the development of a vertebrate organ with critical functions in hematopoiesis, immunity, and blood filtering, the spleen. In humans, spleen agenesis results in a high risk for life-threatening bacterial infections in newborns and children (as in Isolated Congenital Asplenia). While immune functions of the spleen have been well characterized, the specification, morphogenesis, and growth of the splenic anlage, as well as differentiation of the splenic mesenchyme and its cell type composition, remain poorly understood. We have identified novel regulatory networks that control the successive stages of spleen development. In particular, PBX TFs are hierarchical regulators of spleen-specific genetic cascades. Our studies in mouse models enabled the identification of the first genetic etiology of human Isolated Congenital Asplenia. In collaboration with Jean Laurent Casanova’s lab (Rockefeller University & HHMI), we identified NKX2-5, a gene target of PBX1, as the gene mutated in individuals with congenital asplenia in a family from Africa. More recently, the Casanova lab discovered that RPSA, a ribosomal protein, is mutated in approximately 50% of the children affected by Isolated Congenital Asplenia. We are currently studying the roles of Rpsa and other ribosomal proteins in mammalian spleen development using murine loss-of-function models and primary splenic cells from mouse and human embryos. In parallel, we are using single-cell RNA sequencing to define the composition of the heterogeneous splenic stroma during development. This combined approach is leading us to gain a deeper understanding of the pathogenesis of human congenital asplenia, as well as identifying new genes for a rapid prenatal diagnosis of this rare but deadly birth defect, often discovered only at autopsy.
We have devoted substantial efforts to understand the genetic and transcriptional networks that control the development of a vertebrate organ with critical functions in hematopoiesis, immunity, and blood filtering, the spleen. In humans, spleen agenesis results in a high risk for life-threatening bacterial infections in newborns and children (as in Isolated Congenital Asplenia). While immune functions of the spleen have been well characterized, the specification, morphogenesis, and growth of the splenic anlage, as well as differentiation of the splenic mesenchyme and its cell type composition, remain poorly understood. We have identified novel regulatory networks that control the successive stages of spleen development. In particular, PBX TFs are hierarchical regulators of spleen-specific genetic cascades. Our studies in mouse models enabled the identification of the first genetic etiology of human Isolated Congenital Asplenia. In collaboration with Jean Laurent Casanova’s lab (Rockefeller University & HHMI), we identified NKX2-5, a gene target of PBX1, as the gene mutated in individuals with congenital asplenia in a family from Africa. More recently, the Casanova lab discovered that RPSA, a ribosomal protein, is mutated in approximately 50% of the children affected by Isolated Congenital Asplenia. We are currently studying the roles of Rpsa and other ribosomal proteins in mammalian spleen development using murine loss-of-function models and primary splenic cells from mouse and human embryos. In parallel, we are using single-cell RNA sequencing to define the composition of the heterogeneous splenic stroma during development. This combined approach is leading us to gain a deeper understanding of the pathogenesis of human congenital asplenia, as well as identifying new genes for a rapid prenatal diagnosis of this rare but deadly birth defect, often discovered only at autopsy.